FAQよくあるご質問
よくあるご質問
企業情報
創業はいつですか?
1999年12月17日です。
薬を開発するには、何にどのぐらいの時間がかかるのでしょうか?
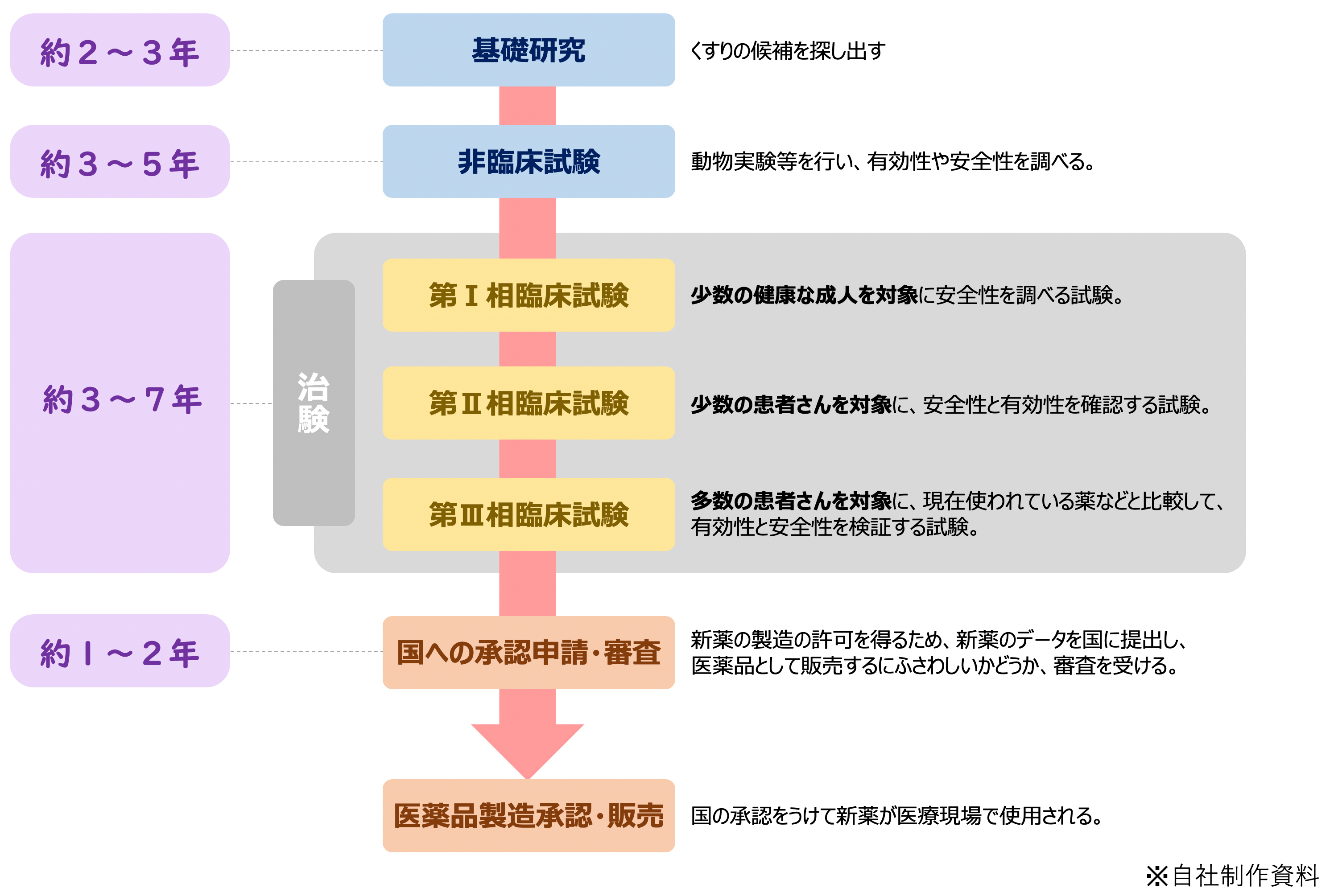
アンジェスの名前の由来は?
社名の「AnGes」は、フランス語で天使を意味する「Ange」に由来し、さらに血管新生(Angiogenesis)や遺伝子制御(Anti-gene)といった医薬品開発に関連する意味も含まれています。
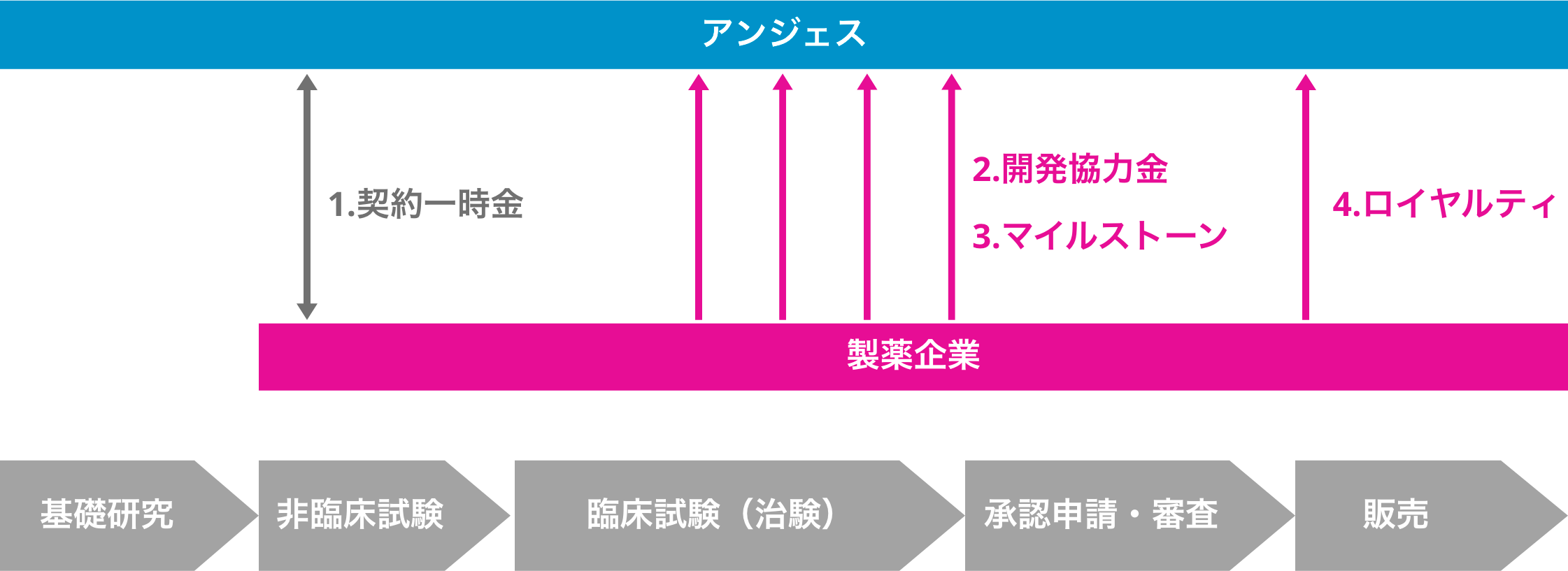
どのようなビジネスモデルですか?
決算・財務関連
決算発表はいつですか?
・第1四半期決算発表は、5月上旬ごろ
・第2四半期決算発表は、8月上旬ごろ
・第3四半期決算発表は、11月上旬ごろ
・決算発表は、2月上旬ごろ に行っています。
決算期はいつですか?
12月です。
決算資料はどこで確認できますか?
下記ページをご覧ください。
過去の業績はどこで確認できますか?
下記ページをご覧ください。
株式情報
株式が上場されたのはいつですか?
2002年9月に、東京証券取引所マザーズに上場しました。
上場取引所はどこですか?
東京証券取引所グロースです。
證券コードは何ですか?
4563です。
取引の単位(1単位)は何株ですか?
100株です。
配当はありますか?
現在はございません。
株主優待制度はありますか?
毎年6月30日及び12月31日現在の当社株主名簿に記載された1単元株式(100株)以上を保有されている株主様を対象として、当社が指定する商品を特別価格で購入できる「株主優待専用申込書」をご所有株式数に応じて年2回お送りしています。
詳しくは、下記ページをご覧ください。
株式の諸手続きについての問い合わせ先はどこですか?
下記ページをご覧ください。
株主関連
株主総会はいつですか?
毎年3月に大阪で開催しています。
議決権行使をするためにはどうすればいいですか?
株主総会基準日(12月末日)時点の株主名簿に記載されている株主様に対して、株主総会の招集通知と議決権行使書をお送りしております。
議決権の行使の方法等は、招集通知でご案内しております。
その他企業全般
IRに関する問い合わせ先はどこですか?
下記ページから受け付けております。なお、多くいただきましたご質問には、当ホームページ上のブログやFAQなどで公開させていただきます。
事業関連
HGF遺伝子治療用製品
HGFとはなんですか?
肝細胞増殖因子(Hepatocyte Growth Factor)で肝臓の細胞を増やす働きをもったタンパクとして発見されましたが、大阪大学の研究室が肝細胞だけでなく血管を新生作用もあることを見出しました。
重症虚血肢とはなんですか?
慢性動脈閉塞症(足の動脈が狭くなったり、詰まったりする病気)のうち、足の安静時疼痛や潰瘍および壊死などを伴う状態のことです。
その他血管疾患との関連性は、こちらのブログをご参照ください。
閉塞性動脈硬化症とはなんですか?
血管疾患に対するグローバルの新しいガイドラインとは?
治療介入をすべき病態を下肢切断のリスクから分類し病態に応じた積極的治療を推奨しているGVG™(Global Vascular Guidelines)になります。
合わせて、こちらのブログもご参照ください。
FDAとはなんですか?
アメリカ食品医薬品局(Food and Drug Administration)の略称で、医薬品や食品などを取り締まるアメリカ合衆国の政府機関です。例えると日本の厚生労働省に似た役割を持っています。
FDAは、消費者が通常の生活を行う際に接する機会がある様々な製品(食品、医薬品、動物薬、化粧品、医療機器、玩具など)の安全性・有効性を確保するための機関です。
適応追加の安静時疼痛に関する進捗状況を教えてください。
2022年9月7日をもって、HGF遺伝子治療用製品の安静時疼痛の適応追加に向けた国内開発を中止いたしました。
NF-κBデコイオリゴDNA
NF-κBデコイオリゴDNAとはなんですか?
どのような症状向けに開発していますか?
椎間板性腰痛症を対象としています。
新型コロナウイルス感染症DNAワクチン
新型コロナウイルス感染症向けDNAワクチンの進捗状況を教えてください。
これまで開発していたDNAワクチンの開発については、2022年9月7日のリリースのとおり、開発を中止いたしました。
その後プラスミドの発現効率や導入効率の向上などが期待される経鼻投与製剤の共同研究を実施いたしました。
Tie2受容体アゴニスト(AV-001)
Tie2受容体アゴニスト「AV-001」とはなんですか?
カナダのバイオ医薬品企業であるVasomune(バソミューン)社と共同開発を進めている急性呼吸不全など血管の不全を原因とする疾患を対象とした医薬品です。中等度から重度のCOVID-19肺炎患者の治療薬として、2020年より米国で臨床試験を開始しています。
Vasomune社とは、どのような会社ですか?
病気から体を守る能力を高める次世代の医薬品を開発するカナダのトロントのバイオ医薬品企業です。Vasomune社は2006年に設立され、血管の正常化戦略に焦点を当てた新しい治療法を用いて医薬品を発見し、開発しています。血管機能障害は、COVID-19、インフルエンザ関連ARDS、急性肺損傷、急性腎損傷、出血性ショック、敗血症、脳卒中など、いくつかの疾患状態の病態と関連しています。
「AV-001」承認のおおよその時期を教えてください。
米国の規制当局であるFDA(アメリカ食品医薬品局)等と協議しながら、なるべく速やかに承認を取得できるよう開発を進めてまいりたいと考えております。
「AV-001」は、前期第II相臨床試験の結果を開示する予定はありますか?
後期第II相臨床試験を開始する時は、前期第II相臨床試験の結果をなんらかの形で開示する前提で進めることになります。
ゲノム編集
ゲノム編集とはなんですか?
ゲノムとはすべての生き物の細胞の中には、DNAという長い分子があります。これは「ATGC」という4種類の文字が30億個以上並んだ「設計図」のようなもので、その生き物の体の特徴すべてが記されています。この設計図全体をゲノムと呼びます。
ゲノム編集は、この長大な設計図の特定の場所だけを狙って切ったり書き換えたりする技術です。
Emendo社とは、どのような会社ですか?
2015年12月に設立したイスラエルに研究拠点を保有する米国の企業です。
独自のヌクレアーゼ(酵素)を用いてゲノム編集する技術の開発を行っています。
2020年12月に当社の子会社となりました。
オフターゲット効果とはなんですか?
オフターゲット効果とは、標的配列(ターゲット配列)に似た配列を持つ、ターゲットではない部分の遺伝子を切断してしまうこと。
オフターゲット効果を低減させるために、
・ゲノム内に標的配列に類似した配列がないか検索
・類似配列が存在する標的は避け、別の標的配列を探すことが求められています。
Emendo社の技術の優位性はどんな点ですか?
独自のヌクレアーゼを探索・最適化するプラットフォーム技術OMNIプラットフォームを保有しています。AIを利用して効率的に新たなヌクレアーゼを開発しています。
独自に開発したOMNIヌクレアーゼは、一般的に使用されているクリスパーCas9では使用が困難なゲノムの編集が可能となるなどの特徴があります。
Emendo社の持つ遺伝子編集技術の名称はなんですか?
「OMNI」といい、「OMNIプラットフォーム」「OMNIヌクレアーゼ」などの名称で使用されます。
合わせてこちらのブログもご参照ください。
マイクロバイオーム事業
マイクロバイオームとはなんですか?
マイクロバイオームは、ヒト微生物叢のゲノムとそれが発現する遺伝子群および微生物叢とヒトの相互作用を含む広い概念を表しています。この微生物叢とヒトは共生しており、ヒトの身体は微生物叢との集合体といえます。
近年では生活習慣の変化がマイクロバイオームの生理状態の変化を誘導しそれが各疾患の原因に関係しているとの報告があり、菌を活用して医療やヘルスケアに役立てるための研究が行われています。
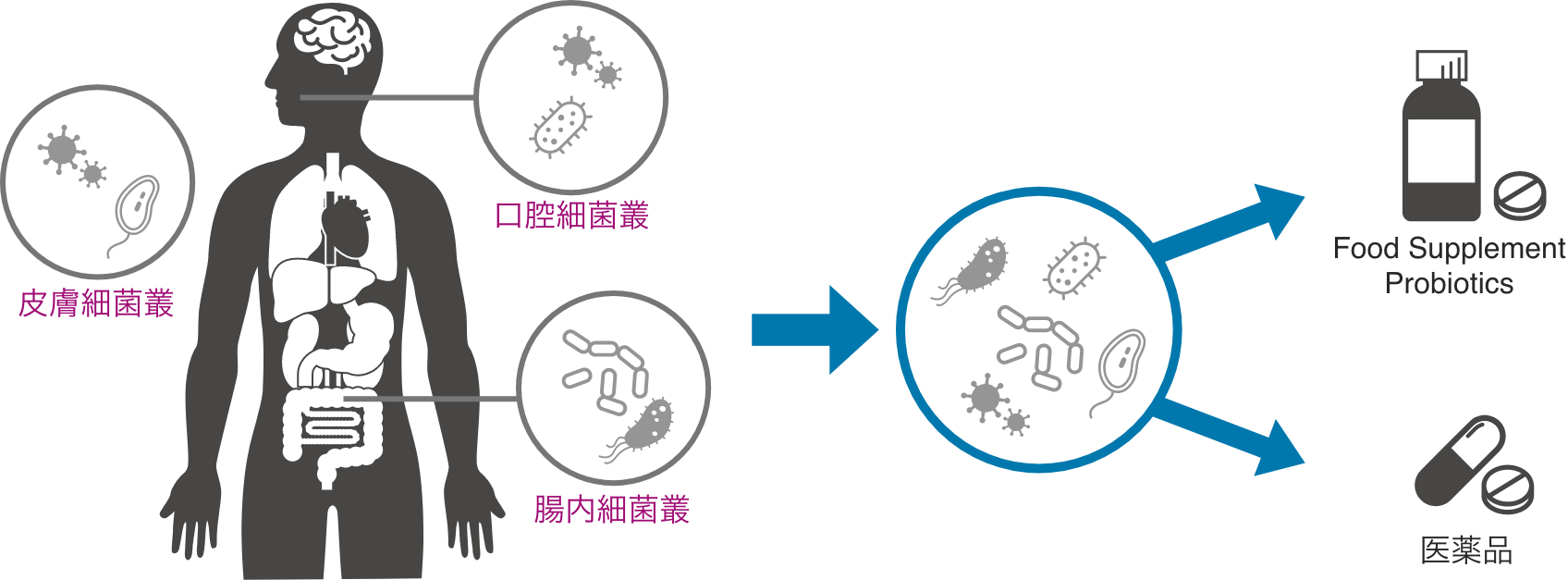
MyBiotics社(MyBiotics Pharma Ltd.)とは、どのような会社ですか?
2014年6月に設立された、マイクロバイオーム領域のパイオニアで、イスラエルのバイオ企業。世界に先駆けて腸内細菌を含む常在菌の培養、製剤化において、その品質や腸内における生存率を向上させる技術を開発しています。これは、マイクロバイオーム医薬の実用化に必須の重要な技術であり、同社はマイクロバイオームの事業化競争において高い優位性を持っています。当社とは2018年7月に資本提携しています。
希少遺伝性疾患のスクリーニング事業関連
アンジェスクリニカルリサーチラボラトリー(ACRL)とは、なんですか?
アンジェスクリニカルリサーチラボラトリー(ACRL)は、希少遺伝性疾患検査を主目的とした衛生検査所*で、2021年4月に開設しました。衛生検査所:人体から採取され又は排出された検体の検査を行う施設で、臨床検査技師等に関する法律に基づき登録された施設。
なぜ、アンジェスクリニカルリサーチラボラトリー(ACRL)を設立したのですか?
希少遺伝性疾患も治療が可能なものが増えていますが、治療が可能であっても、病気の進行を止める治療の場合は、症状が出る前に治療を始めないと、望ましい治療効果が得られないことがあります。また、発症しないと通常の診断では発⾒が難しい疾患もあります。希少遺伝性疾患の治療は、発症早期、望ましくは発症前から開始することが重要です。ACRLでは、全国で無料で実施される新生児のマススクリーニング検査の対象疾患以外の遺伝子の変異が原因で起こる疾患の追加スクリーニング検査を受託しており、希少遺伝性疾患の早期発見に努めております。
希少遺伝性疾患の追加検査とはなんですか?
新生児の遺伝子の変化が原因で起こる疾患(希少遺伝性疾患)のうち、無償で実施される「新⽣児マススクリーニング」に含まれない疾患の可能性を判断するための有償の検査です。
希少遺伝性疾患の追加検査を依頼するには、どうすればいいですか?
各地の対応医療機関にご連絡ください。検査に必要な血液は、原則として「新⽣児マススクリーニング」のための採血時に少量の血液を追加で採取するため、赤ちゃんの負担を最小限にできます。
希少遺伝性疾患の追加検査出来る疾患はなんですか?
ムコ多糖症I型、II型、IVA型、VI型、Ⅶ型、ファブリー病(男児のみ)、ポンペ病、ニーマンピック病A/B型、クラッペ病、副腎白質ジストロフィー(男児のみ)、脊髄性筋萎縮症(SMA)、重症複合免疫不全症、アデノシンデアミナーゼ欠損症の14疾患です。
希少遺伝性疾患の追加検査の検査結果は、どれくらいの期間で分かりますか?
1か月検診時に受診した医療機関で結果をご確認いただけます。
陽性の結果が出たらどうなりますか?
受診した医療機関にご相談ください。
スクリーニング事業の目標はなんですか?
新生児スクリーニングは、希少疾患の発見から治療までを一貫して提供する体制の一部としていきたいと思っています。最大で毎月約1万件の検査に対応できますが、国内で出生する新生児が全て受験できる体制が整うよう関係諸機関と協力して進めたいと考えています。
希少疾患治療薬「ゾキンヴィ」
「ゾキンヴィ」とはなんですか?
早老症といわれる、「ハッチンソン・ギルフォード・プロジェリア症候群(HGPS)」及び「プロセシング不全性のプロジェロイド・ラミノパチー(PL)」を対象疾患とした、希少疾患治療薬です。ゾキンヴィは、HGPS の死亡リスク低減、プロセシング不全性早老性PLの治療薬として、2020年11月に米国で承認されました。当社は、2022年5月に、米国の製薬企業Eiger BioPharmaceuticals Inc. と、「ゾキンヴィ」について独占販売契約を締結しました。2023年3月に厚生労働省により希少疾病医薬品(オーファン・ドラッグ)の指定を受け、2023年5⽉に「HGPS及びプロセシング不全性のPL」を効能又は効果として承認申請しました。2024年1月に製造販売承認を取得し、2024年4月に薬価基準に掲載され、2024年5月27日に販売を開始いたしました。
「ハッチンソン・ギルフォード・プロジェリア症候群(HGPS)」と「プロジェロイド・ラミノパチー(PL)」とは どのような病気ですか?
新生児期ないし幼年期に発症して、全身の老化が異常な速度で進行する早老症疾患です。 身長、体重の発育が乏しく、強皮症などの皮膚老化、脱毛、骨格・歯の形成不良をもたらします。 また、重篤な心機能障害や脳血管障害を招きやすく、平均寿命は約14.5年です。
ゾキンヴィの有効性・安全性はどうなのですか?
ゾキンヴィは、ハッチンソン・ギルフォード・プロジェリア症候群(HGPS)の患者において、死亡率を72%減少させ、平均生存期間を4.3年延長させるというデータがあります。 また、安全性については、多くのプロジェリア患者が10年以上にわたってゾキンヴィ治療を継続しており、 報告された副作用はそのほとんどが軽度または中等度のものです。
ゾキンヴィについてアンジェスの役割はどのようなものですか?
日本国内において承認取得手続きを行い、厚生労働省より日本国内の製造販売承認を取得し、販売をしています。
当社の事業目的である「治療法がない疾病分野や難病、希少疾患などを対象にした革新的な医薬品の開発を通じて、国民生活や医療水準の向上に貢献すること」に合致した製品で、今後も日本のドラッグ・ロス解消に貢献したいと考えています。
なぜ、アンジェスがゾキンヴィの取り組みを始めたのですか?
当社は、「治療法が存在しない難病や希少疾患に挑むことで、世界中の患者様に新たな希望を届けること」を使命としており、ゾキンヴィはこの目標に合致するためです。
また、当社が2021年からアンジェスクリニカルリサーチラボラトリー(ACRL)において実施しているスクリーング検査(希少遺伝性疾患検査事業)とともに、2024年から遺伝学的検査も開始し、ゾキンヴィの販売に合わせHGPSとプロセシング不全性PLの検査も受託できる体制を構築しました。当社は、日本におけるドラッグ・ラグ、ドラッグ・ロスの解消に向け取り組んでまいります。